多形性黄色瘤型星形细胞瘤出现自发性颅内出血检出NTRK融合和CDKN2A纯合缺失_cdkn2a纯合缺失
多形性黄色瘤型星形细胞瘤(PXA)是一种罕见的脑肿瘤,占所有胶质瘤的 1% 不到。由于全球PXA数量有限,对其分子特征的深入了解仍在进行中。另外,自发性颅内出血(pICH)是一种罕见但可能对幼儿造成严重影响,通常由血管畸形或潜在的血液病引起。本文描述了一个有趣的案例,一名幼儿出现pICH,后来发现PXA是出血的潜在原因。对肿瘤进行进一步的分子检测发现,NTRK基因融合和CDKN2A缺失在婴儿高级别胶质瘤中更常见。本文讨论了该病例不寻常的临床病理学特征,并与已发表的文献进行了验证。一名之前健康的 2 岁男性因右额顶叶大面积脑内血肿而出现急性嗜睡和颅内压升高症状。为挽救生命,他接受了紧急开颅手术和部分血肿清除术。后续神经影像学检查报告右侧脑内肿瘤可能存在出血。组织学证实肿瘤为PXA(WHO 2级)。进一步的分子检查显示BRAF V600E突变阴性,但发现了CDKN2A纯合缺失和独特的神经营养酪氨酸受体激酶(NTRK)基因融合阳性。患者随后接受二期手术,最大程度安全切除残留肿瘤,然后开始辅助化疗。迄今为止,只有极少数PXA儿童病例表现出自发性pICH,并接受了详细的分子检测。本文患者的病程凸显了多学科神经肿瘤学团队在指导最佳治疗方面的作用。
胶质瘤是儿童中最常见的原发性中枢神经系统(CNS)肿瘤。其治疗难点往往在于其临床行为广泛。在这一肿瘤中,多形性黄色瘤型星形细胞瘤(PXA)是一种罕见亚型,占所有胶质瘤的 1% 不到。受影响的患者通常表现为具有癫痫发作的青少年或年轻人。世界卫生组织(WHO)最新分类将PXA的分级指定为WHO CNS 2 级肿瘤,因为它具有相对较好的预后,但比其它儿童低级别胶质瘤(LGG)更容易复发。此外,多达三分之一的胶质瘤表现出间变性(WHO CNS 3 级),其特征是有丝分裂活动增加,有时出现坏死,这与总体生存率降低有关。迄今为止,由于全球PXA数量有限,对其分子组成的深入了解仍在进行中。另外,自发性颅内出血(pICH)是一种罕见但可能造成毁灭性后果的儿童急症。特别是在幼儿中,此类病例通常面临临床和诊断方面的挑战。广义上讲,pICH主要与颅内血管异常有关。其它病因,如血液学、全身性和心脏原因、脑肿瘤和颅内感染相对较少见。本文描述了一个有趣的病例,一名 2 岁男性出现危及生命的pICH,最终发现PXA是其出血的根本原因。对肿瘤的进一步分子检测显示,BRAF V600E突变呈阴性,CDKN2A纯合缺失呈阳性。此外,还发现该肿瘤携带NTRK基因融合,这种基因融合在婴儿高级别胶质瘤(HGG)中更为常见。本文讨论了该病例不寻常的临床病理学特征,并与已发表的文献进行了验证。
患儿男,2 岁,既往健康,因在 1 天内出现呕吐、嗜睡和瞳孔不等大症状就诊。就诊前没有外伤史、近期感染史或家族史,因此无法解释患儿的临床表现。紧急脑部CT扫描显示右额顶区有一个大的、急性pICH,伴有脑室内扩散、早期脑积水和右侧钩回疝。为挽救生命,患儿接受了紧急开颅手术并进行了部分血凝块清除。由于诊断结果不寻常,将清除的血肿送去进行病理组织学检测。随后的血液检查排除了潜在的血液学、感染和全身性原因,结果未发现异常。在患者病情稳定后,安排了随访磁共振成像(MRI)。报告称右额顶区病变边界不清,混有血液产物和周围水肿。未见明显颅内血管异常。附加扩散张量成像(DTI)显示残留病变已侵入右侧皮质脊髓束。没有脊髓转移的影像学证据(图1)。
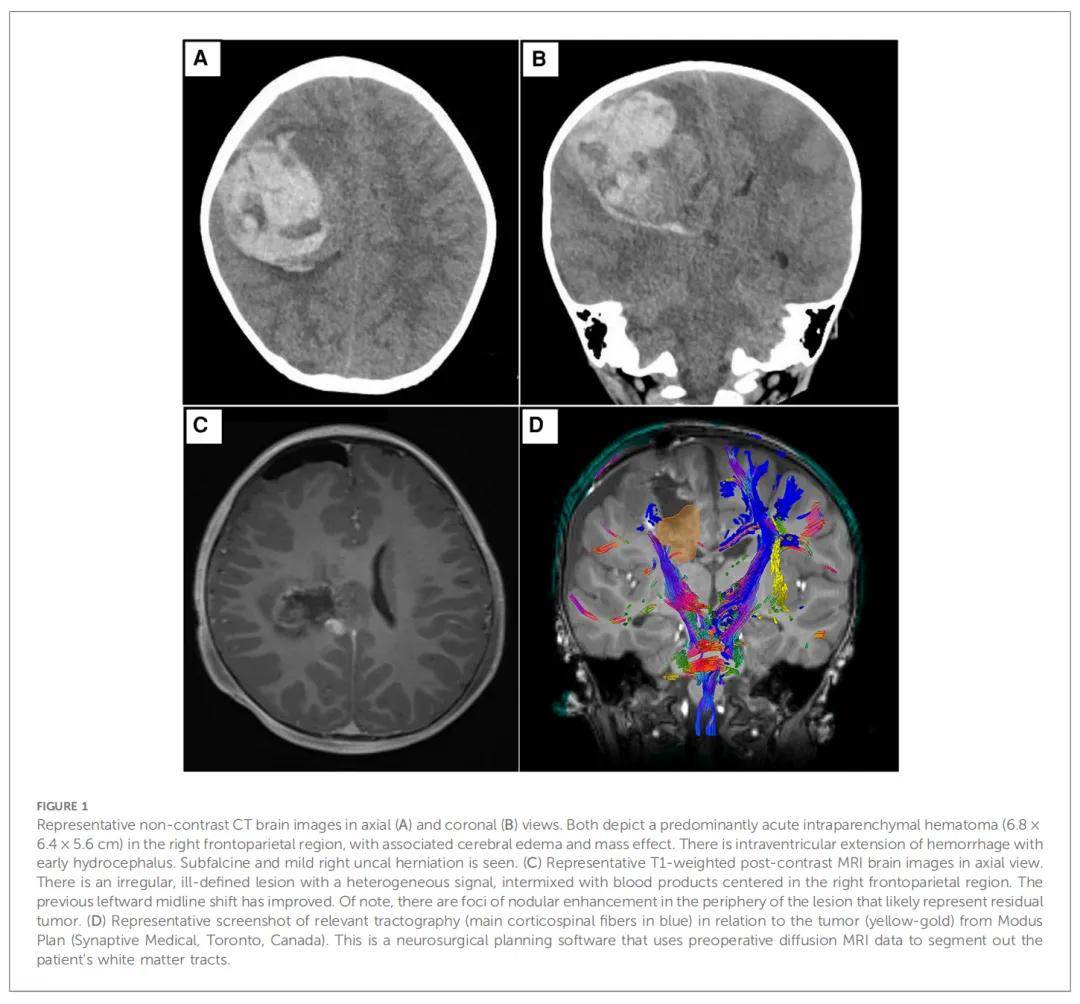
作为综合诊断工作流程的一部分,研究人员会根据患者的临床和影像学结果仔细审核每个肿瘤的初始组织病理学细节。接下来,根据这些发现选择相应的分子检测。在本文病例中,清除血肿的组织病理学报告显示存在边界清晰的内在胶质肿瘤。有丝分裂图为 1 个有丝分裂图/mm2,Ki-67指数高达 5%。值得注意的是,BRAF V600E的免疫组织化学染色为阴性。图2和图3描述了发现的其他说明性细节。此时的累积特征导致初步诊断为WHO CNS 2 级PXA。由于在检查过程中ALK和pan-TRK均呈明显阳性(图2E、F),因此通过NGS进行了检测,结果显示存在ETV6::NTRK基因融合(图3A)。随后使用了另一个NGS panel进行检测,证实了ETV6::NTRK融合的存在。此外,结果显示没有可报告的单核苷酸变异(特别是BRAF V600E)或拷贝数变异。有趣的是,研究人员注意到,两种NGS panel均未检测到ALK基因突变或融合。在这种情况下,研究人员决定与NGS的结果保持一致。但是,同时检测到了细胞周期蛋白依赖性激酶抑制剂2A(CDKN2A)的纯合缺失。鉴于CDKN2A纯合缺失的检出,研究人员安排了CDKN2A基因的荧光原位杂交(FISH)检测,结果确定肿瘤中存在该基因缺失。综合上述结果,最终诊断为“WHO CNS 2 级PXA,伴有ETV6::NTRK融合”。患者在初次手术后约 2 周接受了二期切除术,以切除残留的出血性肿瘤。术中,研究人员决定留下一小块浸润到皮质脊髓束的肿瘤,以避免神经损伤。除此之外,患儿恢复良好,昏迷评分完全正常,没有残留神经功能缺损。第二次手术提交的肿瘤标本在组织病理学上与患者初次手术相似。
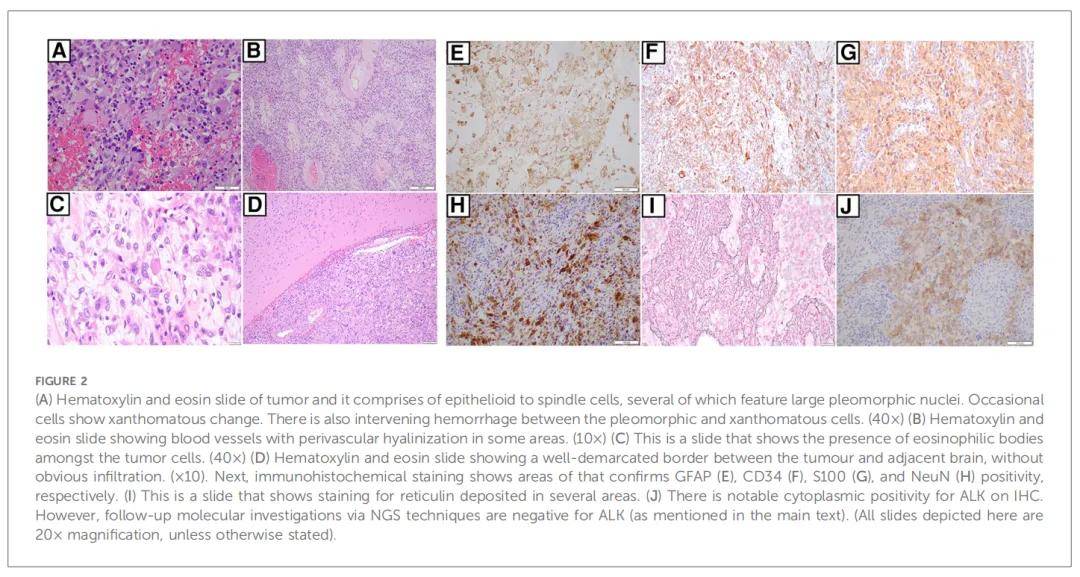
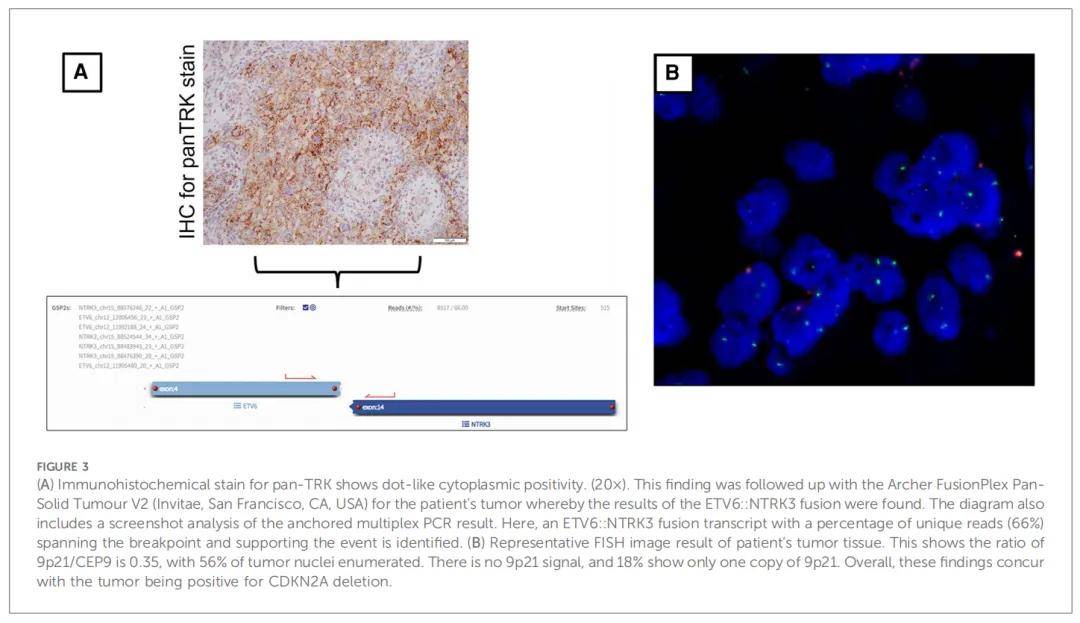
该病例在神经肿瘤多学科肿瘤(MDT)委员会上进行了报告。MDT由来自儿童肿瘤科、神经影像科、病理科、神经外科、肿瘤影像科和相关医疗团队的专家组成。具体针对该患者,讨论了以下几点:首先,大脑重要区域仍有残留肿瘤;其次,Ki-67指数为 5%;最后,分子学发现包括:存在NTRK基因融合、BRAF V600E突变阴性和CDKN2A纯合缺失。共识是按照婴儿HGG进行诊断。尽管在会诊期间进行了多次讨论,但患者的父母拒绝为患者进行推荐的胚系检测。随后,患者开始接受以卡铂为基础的化疗方案,如果复发,可选择使用NTRK抑制剂。鉴于患者年纪尚小,质子束治疗的作用应尽可能推迟。同时,患者被转诊至肿瘤遗传学团队,讨论胚系检测的作用。然而,患儿父母在咨询后拒绝继续治疗。在初次就诊后约 10 个月,患者临床状况良好,没有肿瘤复发的影像学证据。到目前为止,患儿仍很活跃,没有神经系统缺陷或明显的发育迟缓。
PXA于 1979 年首次被描述为一种不同类型的胶质瘤,推测其起源于软脑膜下星形胶质细胞。目前,Kepes等人最初描述的一种幕上星形细胞瘤仍然具有现实意义,该瘤位于皮质表层,具有独特的组织学特征,例如明显的细胞多形性、丰富的网状蛋白网络和明显的含脂质神经胶质细胞。PXA往往位于幕上区域,尤其是颞叶。有时,软脑膜和大脑浅表区域在解剖学上都会受累。偶尔,文献中也会描述幕下病例。在神经影像学上,PXA通常位于周围神经且常呈囊性,涉及大脑皮质和软脑膜。人口统计学上,关于性别偏好的报道相互矛盾,一些论文报告称男女发病率相同,而另一些论文则声称男性略多。研究报告称,PXA通常在 20 岁左右时诊断出来。婴儿HGG组(此处指 3-5 岁以下儿童)发病率不高。至于临床表现,患儿往往表现为癫痫发作。神经外科干预,尤其是肿瘤全切除术,已被证明与良好的生存结果相关。尽管PXA被认为具有相对较好的预后,但与其它儿科LGG相比,其复发风险较高。这种略微更具侵袭性的行为导致其被认定为WHO 2 级或 3 级中枢神经系统肿瘤。现有文献显示,多达三分之一的PXA肿瘤表现出间变性特征,如有丝分裂活动增加和坏死,而这两者都与总体生存率降低有关。这个特殊的亚组被称为PXA WHO 3 级。这些肿瘤更具侵袭性,据报道可通过脑脊液(CSF)扩散,通常发生在疾病复发或恶性转化的情况下。对于PXA WHO 2 级肿瘤,Ki-67标记指数通常 1%,而在其 WHO 3 级中高达 15%。
随着检测技术的进步,为儿童胶质瘤的分子频谱提供了更多有价值的见解。如今,临床已经知晓相当一部分PXA病例存在BRAF V600E基因突变和/或CDKN2A基因变异。此外,Mistry等人对转化为继发性HGG的儿童LGG亚组进行了深入研究,结果表明BRAF V600E突变和CDKN2A缺失构成了临床上不同的继发性HGG亚型。他们报告称,在未显示恶性转化的儿童LGG队列中,BRAF和CDKN2A基因变异较少见,并且BRAF突变患者继发HGG的潜伏期更长。值得注意的是,本文患者的肿瘤中存在CDKN2A缺失。CDKN2A是一种肿瘤抑制基因,可编码p16INK4a蛋白并作为细胞周期进程的抑制剂。分子生物学研究表明,它是许多人类癌症中突变的主要靶点。先前的研究表明,无论组织学分级如何,CDKN2A纯合缺失都是IDH突变型胶质瘤患者生存结果的重要预后因素。例如,在成人WHO 3 级少突胶质细胞瘤中发现CDKN2A基因纯合缺失与较低的生存率相关,无论有无坏死伴有微血管增生。此外,在组织学上较低级别的成人胶质瘤中,CDKN2A纯合缺失与更具侵袭性的临床病程相关,并且是最新WHO分类中 4 级状态的分子标记。有趣的是,最近对 67 例PXA肿瘤的研究报告称,其中高达 94% 的肿瘤有预先存在的CDKN2A/B缺失。然而,进一步的分析表明,这种基因变异与总生存率无关。相反,WHO对PXA的分级是研究队列中生存率的更强预测指标。总体而言,肿瘤的分子信息非常重要,因为针对具有挑战性的癌症(尤其是脑肿瘤)的靶向治疗模式正在发生转变。例如,BRAF抑制剂在携带BRAF V600E突变的肿瘤中获得的BRAF抑制剂用于疾病控制的初步证据是乐观的。同样,细胞周期蛋白依赖性激酶抑制剂(CDK)基因变异的鉴定为开发针对各种恶性肿瘤(包括神经胶质瘤)的CDK相关疗法铺平了道路。
随后,据报道,其余没有BRAF V600E的PXA病例(包括本文患者)患有RTK(受体酪氨酸激酶)基因融合,而不是MAPK(丝裂原活化蛋白激酶)变异。在撰写本文时,文献中只有另外一例儿童PXA病例报告了NTRK融合。广义上讲,基因融合是由于易位、缺失、重复或倒位造成的基因组重排,导致基因之间两个或多个编码调控DNA序列混合而产生的特征性突变。基因融合具有临床意义,因为它们提供了有关肿瘤发生的重要信息,为开发针对特定融合患者的靶向治疗铺平了道路。近年来,转录组分析发现了一组携带基因融合的胶质瘤患者。据报道,对于患病患者,在高级别胶质瘤中基因融合发生率可高达 30%–50%。迄今为止,合作基因组研究表明,婴儿型HGG包含分子上不同的亚群,这些亚群以基因融合为特征。例子包括NTRK融合,它被描述为多种人类肿瘤(包括儿童胶质瘤)的致癌驱动因素。NTRK家族包括NTRK1、NTRK2和NTRK3,它们编码神经营养因子受体原肌球蛋白相关激酶(TRK)组,例如TrkA、TrkB和TrkC。涉及NTRK家族的基因融合是致癌TRK激活的最常见机制之一。这具有临床意义,因为NTRK融合代表了药物可靶向的基因组改变。因此,潜在的 NTRK 靶向治疗为治疗选择有限的非常年轻的脑肿瘤患者带来了希望。到目前为止,NTRK抑制剂目前正在临床试验中用于治疗原发性中枢神经系统肿瘤,并取得了令人鼓舞的结果。本文患儿如果常规治疗失败,后续可以选择使用NTRK抑制剂。
综上所述,本文患者的相关分子学发现总结如下:BRAF V600E突变阴性、存在CDKN2A纯合缺失和ETV6::NTRK基因融合。基于目前对这种罕见脑肿瘤的理解,很难基于分子结果对本文患者的预后影响得出明确结论。然而,PXA肿瘤经常复发,并且与儿童和年轻人中的其它LGG相比,其生存率较低。此外,与其它RAS/MAPK驱动的LGG相比,PXA中更易出现恶性进展。
鉴于本文患者临床表现较为罕见,下面的讨论中将概述儿童非创伤性颅内出血。儿童非创伤性颅内出血(pICH)是指发生在 29 天至 18 岁之间的脑实质出血,无论是否伴有脑室内扩散。通常,pICH在儿童中的发病率极低。由于目前的证据主要依赖于病例报告和小规模病例系列研究,因此确切的发病率尚不清楚。尽管pICH罕见,但它是导致死亡和不可逆神经损伤的重要原因。延迟诊断很常见,因为非常年幼的儿童难以准确表达不适,并且出血可表现为非特异性症状,例如烦躁、嗜睡或头痛。此外,与成人相比,儿童患者中报告的侧化神经系统症状较少。无论如何,治疗应及时、彻底地进行诊断检查,以指导最佳临床治疗。
值得注意的是,儿童pICH病因与成年人群不同,主要以脑血管病变、血液系统疾病、肿瘤和全身性疾病为主。此外,文献显示,pICH可继发于各种类型的肿瘤,包括良性和恶性肿瘤。肿瘤侵蚀皮质动脉、薄壁肿瘤血管或肿瘤内未知的微血管异常被认为是导致出血的因素。具体到PXA,文献中仅有 3 例其他儿科病例表现为颅内出血(表1)。

该病例的独特之处凸显了各专科团队面临的挑战,并强调了采用综合多学科方法进行患者护理的重要性。尽管血管异常是导致pICH的大部分潜在原因,但临床医生需要注意在急性发作期间将其用于脑肿瘤的鉴别诊断。从肿瘤诊断的角度来看,分子检测提供了有价值的信息来指导辅助治疗。本病例报告增加了关于这种罕见原发性脑肿瘤的文献数据。展望未来,研究人员提倡在国际层面开展合作临床和更为深入的分子研究。





